20 Gennaio 2025 
Di Francesco Panico, medico nefrologo
“La ricerca scientifica, benché guidata esclusivamente dal ragionamento, resta pur sempre un’avventura”
Louis Victor Pierre Raymond de Broglie, 1980
PREMESSA
Il rene policistico autosomico dominante dell’adulto o ADPKD (Autosomal Dominant Polycystic Kidney Disease) si distingue da quello autosomico recessivoinfantile o ARPKD (Autosomal Recessive Polycystic Kidney Disease) dovuto alla mutazione del gene PKHD1 (Polycystic Kidney and Hepatic Disease 1) il cui locus è sul cromosoma 6 e la patologia si trasmette solo quando si eredita la copia mutata da entrambi i genitori.
I genitori dei soggetti con ARPKD non sono affetti dalla malattia perché posseggono anche una copia sana del gene PKHD1, sono cioè “portatori sani” e la frequenza è di 1/70; mentre se tutti e due i genitori sono portatori della mutazione genica, il rischio di ereditare un gene malato da entrambi i genitori arriva a 1/4 ovvero il 25%.
L’ARPKD èuna patologia con carattere fibrocistico epato-renale caratterizzata da dilatazione cistica e ectasia dei tubuli collettori renali con malformazione del piatto duttale epatico e fibrosi epatica o Congenital Hepatic Fibrosis (CHF) con oligoidramnios, ipoplasia polmonare, epicanto oculare e Malattia Renale Cronica (CKD) neonatale.
Questa patologia ha un’incidenza di 1/20.000-40.000 nati vivi e non sempre colpisce fegato e rene.
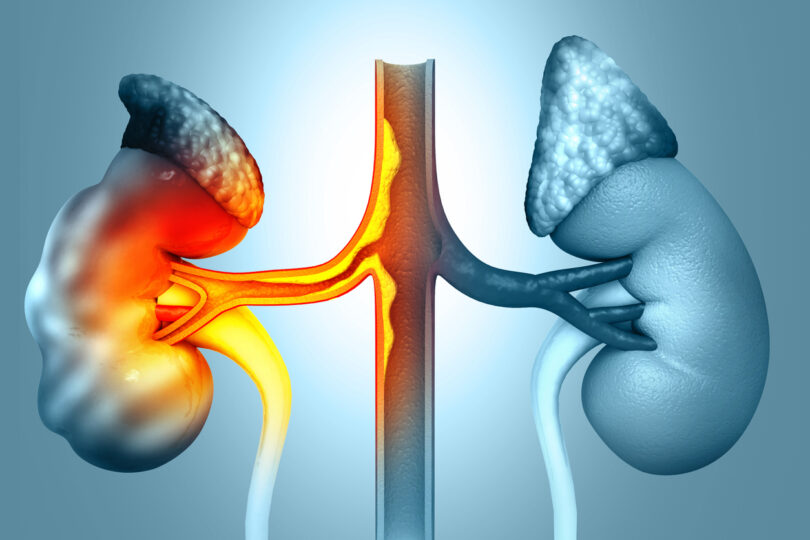
L’ADPKD, al contrario è una patologia monogenica-ereditaria dominante, cioè è necessaria una sola mutazione allelica per il manifestarsi della patologia e ha un’incidenza di 1/400-1.000 nati vivi. L’ADPKD dopo anni dall’esordio sfocia nell’insufficienza renale terminale o End Stage Renal Disease (ESRD) nel 75% dei pazienti prima dei 70 anni e dal momento che la diffusione o prevalenza nella popolazione non supera i 5/10.000 nati, è definita “Rare Disease”.
La caratteristica principale dell’ADPKD è la lenta e costante crescita delle cisti renali con ingombro addominale, dolore, ipertensione arteriosa, ematuria copiosa per rottura delle cisti e/o microematuria con emorragie intra-cistiche, calcolosi e raramente anemia.
L’iperfiltrazione glomerulare per anni sostiene la funzione renale (FR) e solo dopo il totale sovvertimento del parenchima renale si ha CKD e ESRD.
In conclusione l’ADPKD è una patologia renale dal lungo decorso e come vedremo abbastanza responsiva alla terapia medica.
L’ADPKD è dovuta alla mutazione di 2 geni:
- PKD-1 sul cromosoma 16 nell’85% dei casi (Europa)
- PKD-2 sul cromosoma 4 nel 15% dei casi prevalentemente Nord America (USA e Canada).
I soggetti con mutazione del gene PKD-2 tendono ad avere patologia renale più lieve, minor numero di cisti e esordio dell’ESRD più lento al contrario dei soggetti con mutazione del gene PKD-1.
Numerosi studi di biologia molecolare in vivo hanno dimostrato il ruolo della Policistina-1, glicoproteina della parete cistica.
La Policistina-1 derivata dal gene PKD-1 svolge un ruolo centrale nella formazione dei tubuli renali, mentre la Policistina-2 derivata dal gene PKD-2 sembra essere implicata nella formazione dei canali ionici. Inoltre è emerso che la Policistina-1 interagisce con la Policistina-2 regolandone le funzioni e se queste due proteine sono alterate si ha la formazione di cisti.
Uno dei punti deboli, se così può dirsi, dell’ADPKD è che la mutazione si verifica su una sola copia del gene associato alla malattia, e non nell’altro per cui il gruppo di Vishal Patel dell’University of Texas Southwestern Medical Center, ha sperimentato un approccio terapeutico sullacopia sana conla tecnicadel CRISPR Cas-9 (Nature),aprendo nuovi orizzonti alla possibile terapia genica dell’ADPKD come lo è per diverse neoplasie e malattie genetiche: Fenilchetonuria e Amiloidosi ATTR.
DIAGNOSI DI ADPKD
La comprensione dell’ADPKD così come per altre malattie geneticamente trasmesse, è stata possibile grazie al lavoro dell’abate, biologo e matematico ceco Gregor Johann Mendel con l’enunciazione delle leggi della trasmissione ereditaria, osservando le varietà fenotipiche dei piselli che coltivava e incrociava tra loro.
LEGGI DELLA TRASMISSIONE DEI CARATTERI EREDITARI (G.J.MENDEL, 1866)
- 1^ Legge o Dominanza dei caratteri
- 2^ Legge o Segregazione dei caratteri
- 3^ Legge o Assortimento indipendente dei caratteri
L’ADPKD è una patologia genetica dominante con il 50% di probabilità di trasmettersi nei soggetti a rischio, ovvero nei consanguinei di I° di soggetti con mutazione genica.
La diagnosi oggi è possibile con l’Ultrasuonografia (US) o Ecografia Renale, e dato che la presenza di cisti semplici renali è di comune riscontro, Kenneth D. Gardner Jr. già nel 1975 affermò che: “Voler distinguere una malattia cistica renale dalle cisti è come voler distinguere un formaggio svizzero dai buchi”.
Attualmente sono stati codificati “Criteri Unificati” per la diagnosi e/o l’esclusione di ADPKD:
- Presenza di 3 o più cisti renali in un soggetto a rischio con età fra 15-39 anni
- Presenza di 2 o più cisti per rene in un soggetto a rischio con età tra 40-60 anni sono un criterio sufficientemente valido per la diagnosi di ADPKD, come la totale assenza di cisti renali è valida ad escludere l’ADPKD solo in soggetti a rischio con un’età superiore ai 40 anni e non inferiore ai 40 anni senza informazioni genetiche precise. Nello screening dei donatori per trapianti è usata la RMN o TC con mdc e il “riscontro di meno di 5 cisti renali è un criterio valido per escludere l’ADPKD”.
CYSTIC DISEASES OF THE KIDNEY (Kenneth D. Gardner Jr.)
DISPLASIA RENALE
- Displasia multicistica: unilaterale o bilaterale
- Dislpasia cistica focale o segmentale
- Displasia cistica associata a ostruzione distale del tratto urinario
- Displasia cistica familiare
MALATTIA POLICISTICA
- Mal. Policistica infantile: della prima infanzia, dell’infanzia e fibrosi epatica congenita
- Malattia policistica dell’adulto
CISTI RENALI ASSOCIATE A PATOLOGIE EREDITARIE
- Sindrome di Mekel: microcefalia, encefalocele posteriore, abnormità oculari, genitali, polidattilia, palatoschisi, cisti epatiche giganti.
- Sindrome cerebroepatorenale di Zellweger: emosiderosi epatica, iposviluppo cerebrale, criptorchidismo
- Distrofia toracica di Jeune
- Sclerosi tuberosa complessa o malattia di Hippel von Lindau
- Cisti corticali in sindromi malformative multiple
CISTI CORTICALI
- Malattia cistica glomerulare diffusa
- Microcisti della corticale periferica
- Microcisti della corticale iuxtamidollare
- Cisti semplici, solitarie e multiple
CISTI DELLA MIDOLLARE O DISORDINI CISTICI DELLA MIDOLLARE
- Rene a spugna midollare
- Malattia cistica midollare: Nefronoftisi familiare giovanile, malattia cistica della midollare, displasia reno-retinica
- CISTI EXTRA-RENALI: cisti pielogeniche o diverticoli pelvici, cisti parapieliche o linfangectasie, cisti perirenali
MALATTIE CISTICHE EREDITARIE NEGLI ANIMALI
Anche tra gli animali non è rara la presenza di cisti renali e la prima descrizione è stata possibile grazie agli studi di Schlumberger (1950) con la scoperta di cisti renali nel Carassium Auratus (Gold fish) e nelle carpe: cisti renali a carico della capsula del Bowmann, dei tubuli prossimali e neurofibromatosi cutanea.
Nel 1976 Lozzio descrisse in ratti Gunn e Wistar alterazioni cistiche renali che includevano: idronefrosi, macroscopiche cisti renali e tubulari.
Cisti renali sono descritte spesso anche nei gatti e in alcune specie canine, come boxer e dalmata.
DIAGNOSI MOLECOLARE DI ADPKD
Attualmente due sono le tecniche maggiormente utilizzate:
- Analisi mutazionale di Sanger che è in grado di sequenziare gli esoni e le giunzioni dei geni PKD-1 e PKD-2
- Tecnica del linkage soprattutto nello screening embrionario e nella genetica pre-impianto (Pre-implantation genetic diagnostics-PGD).
Il gene PKD-1 è un’unità genica grande e complessa e i primi 33 esoni sono duplicati in 6 pseudo-geni (PKD-1P1 fino a PKD-1P6) che rendono lo screening difficoltoso e complesso ma attualmente facilitato dalla tecnica della PCR (Polymerase Chain Reaction) oppure della MPLA (Multiplex Ligation dependent probe Amplification), metodica quantitativache permette di documentare e identificare gli arrangiamenti genici.
Invece il gene PKD-2 è un gene a copia singola e di facile approccio nello screening mutazionale. Attualmente il test genetico non è richiesto nella maggior parte dei soggetti a rischio, ma può esserlo dopo un’ecografia atipica o ambigua a causa di malattia cistica renale asimmetrica, di PKD lieve o sporadica e in soggetti con sintomatologia tipica ma senza anamnesi familiare positiva.
Sicuramente la diagnostica, con il progresso tecnologico della biologia molecolare, nel prossimo futuro ci sorprenderà e già ora la Next Generation Sequencing (NGS) è capace di rivelare mutazioni complete alleliche e geniche anche a carico di altri cistogeni: geni PKHD1 e HNF1-Beta capaci di modulare la gravità della malattia renale nell’ADPKD.
ADPKD E FUNZIONE RENALE
Nell’ADPKD il GFR è normale per molti anni e per questo non è un marker affidabile, al contrario il monitoraggio del volume renale totale o Total Kidney Volume (TKV) rapportato all’età rappresenta il gold-marker sia primario che secondario nell’evoluzione della CKD.
Il TKV è una stima precisa della massa cistica eben si correlata a diverse manifestazioni cliniche: dolore, ipertensione arteriosa, macro-ematuria e proteinuria, e aumenta in modo esponenziale nella totalità dei soggetti con ADPKD in modo variabile ma unico.
Nel soggetto adulto con ADPKD il volume renale aumenta del 5-6%/anno ben correlandosi all’età e GFR.
Il TKV è studiato con tecniche di imaging:
- ECOGRAFIA O ULTRASONOGRAFIA (US): anche se poco costose, le misurazioni con questa metodica hanno come limite maggiore di essere operatore dipendente quindi poco precise e riproducibili e spesso tendono a sovrastimare il TKV. La misurazione con US del TKV si avvale dell’equazione ellissoidale i cui parametri essenziali sono: la lunghezza ortogonale massima, l’ampiezza e la profondità renale.
- RMN e TAC: sono precise e riproducibili. Le immagini ottenute con la RMN, sia classica che elastografica, possono essere T2 pesate e offrono informazioni circa il volume totale delle cisti e allo stesso tempo escludono l’uso del Gadolinio e di conseguenza il rischio di Fibrosi Sistemica Nefrogenica.
Ambedue le metodiche nella stima del TKV si avvalgono dell’equazione ellissoidale. Oggi in diversi trials si tende a studiare anche la “determinazione del tessuto non cistico” come marker di progressione della malattia e avvalendosi di immagini TAC elaborate dopo mdc, distinguono un “compartimento renale full enhanced” ed “un compartimento intermedio o hypoenhanced”.
Il compartimento hypoenhanced, si caratterizza per un minor incremento del segnale dopo mdc, è in massima parte costituito da tessuto fibrotico e il rapporto volume intermedio/volume parenchimale si correla a variazioni nel tempo del GFR.
- REFERTAZIONE STANDARDIZZATA: deve obbligatoriamente includere:
- Numero preciso delle cisti, ad es. se < a 10 in ogni rene e fegato
- Volume minimo e massimo in ambo gli organi
- Coesistenza di cisti esofitiche o complesse
- Pattern anatomico predominante nella distribuzione delle cisti, ad es. nella corticale, midollare o diffuse.
Quindi Il monitoraggio del TKV sembra possedere tutti i requisiti di un ottimo marcatore prognostico purtroppo allo stato attuale non è una tecnica molto usata nei trials come end-point primario.
- STIMA DELL’Estimated Glomerular Filtration Rate (eGFR) con le equazioni MDRD-4, MDRD-6, CKD-EPI Creatinina, CKD-EPI Cystatina C e CKD-EPI Creatinina-Cystatina C: tutte valide ma con dei limiti rappresentati da: massa muscolare, idratazione, attività fisica, assunzione di farmaci capaci di interferire con la secrezione di creatinina come il Trimetoprim (l’ADPKD è una patologia tubulare), ad eccezione della Cystatina che risente di altri fattori: obesità, stato infiammatorio, alterazioni ormonali tiroidee e glucocorticoidee.
Poco utilizzato nei grandi trials è il GFR misurato (mGFR), determinato sulla base delle clearance plasmatiche o urinarie di marker di filtrazione esogena come l’Inulina o lo Ioexolo, che resta il Gold standard.
- PROTEINURIA: presente in circa il 25% degli adulti con ADPKD eoscilla con valore >300 mg/die e mai > a 1 g/die, ove mai sia presente in range nefrosico bisogna sospettare una diversa patologia renale sovrapposta.
Non è chiaro se la proteinuria sia glomerulare o tubulare, invece è certo che la presenza e il range proteinurico influenzano negativamente il GFR e si associano ad un maggiore TKV.
La terapia anti-proteinurica si avvale di farmaci (ACE Inibitori e Inibitori dell’Angiotensina II) identici a quella della popolazione generale.
- IPERTENSIONE secondaria a varie noxe:
- Aumento dell’attività del sistema Renina-Angiotensina-Aldosterone (RAAS),
- Incremento del tono simpatico
- Problemi meccanici come lo stiramento del peduncolo arterioso renale (Modello Goldblatt).
Lo studio HALT-PKD ha evidenziato che target pressori più bassi di quelli normalmente raccomandati sono risultati più vantaggiosi in pazienti giovani con ADPKD e stadio 1-2 di CKD, non diabetici e senza comorbidità cardiovascolari.
L’ipertensione nell’ADPKD si avvale della stessa terapia della popolazione generale:
- Inibitori del Sistema Renina Angiotensina (RAAS) sono i farmaci di prima scelta. Seguono gli:
- ACE I
- Inibitori del recettore per l’Angiotensina (ARB)
- Inibitori recettoriali dei Mineralcorticoidi (Spironolattone e Eplerenone) soprattutto per il loro effetto anti-fibrosi interstiziale che è cruciale nella progressione dell’ADPKD.
Controverso è l’uso:
- Bloccanti dei canali del calcio o Calcio Antagonisti (VOC-L) perché dimuendo la concentrazione del calcio intracellulare nei dotti collettori inducono un aumento della proliferazione cellulare tubulare e secrezione dei fluidi, tutti momenti che accelerano la crescita delle cisti e il declino del GFR.
Da evitare:
- Diuretici poiché incrementano le concentrazioni sieriche di Arginina Vasopressina (AVP o ADH) favorendo l’ingrossamento e l’aumento delle cisti, e per tale motivo sono da preferire:
- Acquaretici o “Vaptani” antagonisti dei recettori V2 nei dotti collettori con blocco delle Aquaporine 2(AQP-2) e dall’effetto antifibrotico
- SEMPLICI REGOLE DI VITA:
- BMI compreso tra 20-25 Kg/mq,
- Introito salino < 5 g/die di Sodio Cloruro = a 2 g/die di sale,
- Adeguata attività fisica giornaliera.
TERAPIA MEDICA TRADIZIONALE E NUOVI APPROCCI TERAPEUTICI
Molti dei soggetti con ADPKD tra i 50-70 anni presentano ESRD e la terapia in uso in altre forme di CKD: ipotensivanti e diete a ridotto intake proteico, non si sono dimostrate soddisfacenti nel congelare la progressione della CKD, ma allo stesso tempo si raccomanda un intake proteico non > 0,8 g/Kg/die con eGFR > a 30 ml/m’,1,73 mq (KDIGO: Guideline on CKD Evaluation and Management, 2012).
Sulla scorta di nuove ipotesi patogenetiche e fisiologiche dell’ADPKD, si è testato l’impiego di nuovi farmaci:
- INIBITORI DELL’AVP o ADH: che agiscono sui recettori V2 incrementando l’AMPc e bloccano la formazione ex novo o ingrandimento delle cisti. La prova è data dalla diminuzione della Copeptina (frammento C-terminale e precursore della Vasopressina) nell’ADPKD.
Difatti AVP e Copeptina sono rilasciate dallo stesso precursore ormonale in quantità equimolari e per questo motivo la Copeptina è utilizzata come marcatore surrogato dell’AVP essendo molto più stabile dello stesso AVP (Copeptina pro-AVP). I livelli aumentati di Copeptina, sia in studi trasversali (cross-sectional) che longitudinali, si sono dimostrati ottimi marcatori riguardo la gravità e progressione della malattia e per tale motivo si è preso in considerazione l’uso dei Vaptani, che nel trial TEMPO 3-4 su circa 1500 soggetti con ADPKD e eGFR > 60 ml/m’/1,73 mq, hanno evidenziato una riduzione del TKV pari al 48% e un rallentamento del declino del GFR pari al 25%.
Dal 2014 l’agenzia regolatore del farmaco giapponese PMDA (Pharmaceutical and Medical Devices Agency) ha autorizzato l’uso del Tolvaptan (SAMSCA) nella terapia dell’ADPKD, al contrario la FDA e EMA hanno evidenziato delle perplessità:
- TKV non ancora riconosciuto negli USA e Europa come end point primario
- Potenziale azione epatotossica
- Non accettabile il recupero/anno del GFR pari a 1 ml/m’/1,73 mq
L’apporto idrico è stato studiato soprattutto in modelli animali ma non sull’uomo, in quanto non è chiaro né facile da stabilire quale sia l’apporto idrico consigliato sebbene si consigli di mantenere una uOSM/Kg = 250 tenendo sempre presente il rischio di iponatriemia!!
Al momento tra gli studi in corso per frenare la CKD nell’ADPKD desta particolare attenzione l’attivazione della via metabolica m-TOR1 nel tessuto cistico.
In studi su roditori l’impiego di inibitori selettivi della via m-TOR quali Rapamicina e i suoi analoghi: Sirolimus e Everolimus, e la dieta Chetogenica o Cheto-Citra negli stadi 2-3, lasciano ben sperare per il futuro.
Diversi trials hanno dimostrato che l’ADPKD si associa ad anomalie metaboliche quale l’“Effetto Warburg” (18.000 pubblicazioni dal 2000 al 2015!!!) come in oncologia, dove è noto che la maggioranza delle cellule cancerogene produce energia a partire dalla glicolisi seguita dalla fermentazione dell’acido lattico nel citosol piuttosto che da una percentuale relativamente bassa di glicolisi seguita dall’ossidazione del piruvato nei mitocondri come nella maggior parte delle cellule normali.
Diversi studi in vitro e in vivo hanno dimostrato che le cellule epiteliali che rivestono le cisti sono strettamente dipendenti dal glucosio e che un metabolismo chetogenico contrasta la crescita delle cisti con effetti benefici sul eGFR.
A tal proposito è interessante, anche se preliminare, lo studio KETO-ADPKD pubblicato su Cell Report Medicine, in cui sono stati arruolati 66 pazienti divisi in random di 3 gruppi omogenei: il 1° a dieta chetogenica (DK), il 2° a “digiuno con acqua e sali minerali o Water Fasting” (WF) per 3 giorni al mese, il 3° o gruppo di controllo dieta libera.
I dati finali hanno evidenziato che nel 95% dei pazienti del 1° gruppo e nel 85% dei pazienti del 2° gruppo si assisteva ad una riduzione della chetogenesi dimostrata dalla misurazione dell’acetone nel sangue e nel respiro. La sola DK ha comportato una riduzione significativa della massa grassa e del volume epatico e una riduzione statisticamente significativa del volume renale, mentre i pazienti del gruppo di controllo e quelli che in dieta WF al termine dei 90 giorni mostravano un declino progressivo dell’eGFR cosa che non si aveva nel gruppo con DK.
Questo studio offre dati promettenti con le Diete Chetogeniche nell’ADPKD ma occorrono studi più estesi nel tempo e su numero più ampio di pazienti per poter avere un quadro completo sull’efficacia e sicurezza, pertanto questo studio al momento non è una raccomandazione a seguire una DK a discapito di altre terapie più consolidate dalla pratica clinica.
- ANALOGHI SOMATOSTATINA (Lanreotide e Octreotide) sono stati studiati nell’ADPKD (Aladin Trial) monitorando il TKV con dati incoraggianti.
- INIBITORI HMG CoA-Reduttasi (STATINE): Pravastatina soprattutto in bambini con ADPKD con eGFR >80 ml/m’/1,73 mq con ottimi risultati sia per il TKV che eGFR.
- EMATURIA: sia macro che microscopica (Ac.TRANEXAMICO) Antifibrotico
- Emorragie intra-cistiche, complicanza frequente nell’ADPKD (60%)
- Calcolosi
- Infezioni
- Traumatismi addominali
- Carcinomi renali o uroteliali di rado.
L’ematuria può essere asintomatica o esordire sotto forma acuta come colica e ed autolimitantesi in circa 7 gg.
- CALCOLOSI dovuta a:
- Stasi urinaria,
- Ridotto pH,
- Ridotta escrezione di NH4 e di citrato.
La tecnica di imaging più utile è la TC Dual source capace di differenziare la calcolosi uratica da quella da ossalato di calcio. Terapeuticamente il Citrato di potassio è il trattamento elettivo per le condizioni proprie dell’ADPKD: ipocitraturia e deficit di acidificazione distale.
- MONITORAGGIO E TERAPIA DELLE SEPSI CISTICHE SIA RENALI CHE EPATICHE: l’infezione cistica è da considerarsi soprattutto in presenza di febbre con andamento settico, dolore addominale, PCR elevata, con urinocultura ed emocultura negative. L’esame per eccellenza è la PET con 18F-Fluorodesossiglucosio. Terapeuticamente danno buoni risultati gli Antimicrobici liposolubili: Fluorochinolonici e il Trimetopim-sulfametazolo. La certezza dell’avvenuta eradicazione richiede almeno 2 urinoculture negative.
- TERAPIA DEL DOLORE: nell’ADPKD il dolore è una manifestazione tipica e costante ed può essere dovuto: a iperattività neuronale sensitiva del SNA del rene, pelvi e uretere. Terapeuticamente per l’eradicazione del dolore cronico ci si avvale della
- Sclerodermia percutanea delle cisti o la fenestrazione per via laparoscopica
- Blocco con radiofrequenze del plesso celiaco e stimolazione del midollo spinale.
Se la sintomatologia algica è particolarmente disabilitante si può ricorrere alla simpatico-splancnicectomiatoracoscopica gravata però da grosse complicanze: pneumotorace ed grave ipotensione ortostatica. Poco o per nulla usata è la denervazione renale laparoscopica.
- GRAVIDANZA: le donne in età fertile devono essere edotte sul:
- Peggioramento dell’eventuale malattia cistica epatica (Polycystic Liver Disease o PLD) dopo terapiacon estro-progestinici
- Rischio elevato di HPE gestosi soprattutto se anamnesticamente è presente ipertensione pre-gravidica.
- TERAPIA ESRD: il trapianto è la scelta ottimale e nell’attesa l’emodialisi è una scelta terapeuticamente valida anche se la dialisi peritoneale non rappresenta una controindicazione assoluta (Esperienza di Hong Kong dove la DP è il trattamento di scelta nell’ESRD anche in ADPKD).
- TRAPIANTO: in genere nella fase di preparazione pre-trapianto si decide di rimuovere uno dei due reni nativi soprattutto quando sono presenti infezioni gravi e/o ricorrenti, calcolosi sintomatica, sanguinamento grave e/o ricorrente, dolore incoercibile, sospetto di neoplasia e problemi di spazio addominale per allocare il rene da trapiantare. Globalmente la morbilità post-trapianto non sembra essere maggiore nei soggetti con ADPKD rispetto alla popolazione trapiantata. Così come il rischio di sviluppare carcinoma non si discosta statisticamente dai soggetti con altre patologie
- EDITING GENOMIC CRISPR Cas-9 (Clustered Regularly Interspaced Short Palindromic Repeats ovvero Sequenze geniche che si ripetono ad intervalli regolari): l’utilizzo di questa tecnica ha permesso al gruppo di Patel di dimostrare che il gene PDK1 è dotato di un sito di legame (mir-17) specifico per un micro-RNA espresso ad elevati livelli in alcuni modelli della malattia e hanno verificato se il blocco di tale via bloccasse la formazione delle cisti. Questa tecnica permette che qualunque tipo di cellula può essere modificata e corretta anche per un solo errore e ovunque nel genoma. Il CRISPR o modello di sviluppo di editing genomico è stato premiato con il Nobel per la Medicina nel 2020 (E. Charpentier e Jennifer A. Doudna).
A CRISPR sono associati dei geni Cas che codificano enzimi capaci di tagliare il DNA non in modo causale ma in modo preciso grazie a un RNA guida. La strategia di editing basata su CRISPR può utilizzarsi anche in vivo mediante l’inoculo di nanoparticelle lipidiche.
PATOLOGIE EXTRARENALI NELL’ADPKD
- ALTERAZIONI CARDIACHE:
- Prolasso mitralico,
- Versamento pericardico,
- Aneurismi e dissecazione del tratto ascendente dell’aorta, delle arterie spleniche e poplitee
- ALTERAZIONI EXTRACEREBRI:
- Cisti aracnoidee asintomatiche
- Dilatazioni aneurismatiche circolo di Willis
- ALTERAZIONI PANCREATICHE:
- Cisti pancreatiche
- ANOMALIE INTESTINALI:
- Diverticolosi colica soprattutto nell’ ESRD dell’ADPKD
- ALTERAZIONI DELLA PARETE ADDOMINALE:
- Ernie inguinali o paraombelicali, importanti se si opta per la dialisi peritoneale (PD)
- ALTERAZIONI EPATICHE: rara è la
- Fibrosi epatica congenita con ipertensione portale nell’ASPKD (rara)
- Splenomegalia e lobo epatico sinistro aumentato di volume (costanti)
CONCLUSIONI
Gli studi molecolari dell’ultimo decennio sui geni PKD-1 e PKD-2 hanno fatto emergere sostanzialmente due filoni di ricerca riguardo alle alterazioni del rene policistico:
- Le cellule che formano le cisti hanno una crescita esagerata rispetto al rene normale.
- Queste cellule sono attivamente secernenti e ciò provoca la loro espansione.
Grazie a queste conoscenze sono stati avviati studi, anche se al momento su una ristretta coorte di pazienti, utilizzando farmaci e/o tecniche genomiche già in uso per altre patologie rivolte alla correzione di questi due difetti.
BIBLIOGRAFIA CONSULTATA
- Spithoven E, Kramer A, Meijer E: Renal replacement therapy for ADPKD in Europe: prevalence and survival an analysis of data from the ERA-EDTA Registry. Nephrol Dial Transplant 2014; 29: 15-25
- Chapman AB, Bost JE, Torres VE: Kidney Volume and Functional Outcomes in ADPKD. Clin J Am Soc Nephrol 2012; 7: 479-486
- Hateboer N, van Dijk MA, Bogdanova N: Comparison of phenotypes of PKD types 1 and 2. Lancet 1989; 353: 103-107
- Ravine D, Gibson RN, Walker RG: Evaluation of ultrasonographic diagnostic criteria for ADPKD 1824-827. Lancet 1994; 343: 824-827
- Pei Y, Hwang YH, Conklin J: Imaging based diagnosis of ADPKD. J Am Soc Nephrol 2015; 26: 746-753
- Tan AY, Michaeel A, Liu G: Molecular diagnosis of ADPKD using next-generation sequencing. J Mol Diagn 2014; 16: 216-228
- Bergman C: ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocpies. Pediatr Nephrol 2015; 30:15-30
- Collins SC: Preimplantation genetic diagnosis: thecnical advances and expanding applications. Curr Opin Obstet Gynecol 2013; 25:201-206
- Irazabal MV, Rangel LJ, Bergstrahl EJ: Imaging classification of ADPKD: a simple model foe selecting for clinical trials. J AmSoc Nephrol 2014; 1: 160-172
- Torres VE, Chapman AB, Devuyst O: Tolvaptan in patients with ADPKD. N Engl J Med 2012; 367: 2407-2418
- Schrier RS, Abebe KZ, Perrone RD: Angiotensin blockade, blood pressure and ADPKD. N Engl J Med 2014; 371: 2255-2266
- King BF, Reed JE, Bergstralh EJ: Quantification and longitudinal trends of kidney, renal cyst, and renal parenchyma volumes in ADPKD.J Am Soc Nephrol 2000; 11: 1505-1511
- Bakker J, Olree M, Kaatee R: Renal volume measurements: accuracy and repeatability of US compared with that of MR imaging. Radiology 1999; 211: 623-628
- Sise C, Kusaka M, Wetzel LH: Volumetric determination of progression in ADPKD by computed tomography. Kidney Int 2000; 58: 2492- 2501
- Fick-Brosnahan GM, Belz MM, Mcfann KK: Relationsship between renal function in ADPKD: aongitudinal study. Am J Kidney Dis 2002; 39: 1127-1134
- Caroli A, Antiga L, Conti S: Tomography evaluation of ADPKDIntermediate volume on computed tomography imaging defines a fibrotic compartment that predicts GFR decline in ADPKD. Am J Pathol 2011; 179: 619-627
- Antiga L, Piccinelli M, Fasolini G: Computed tomography evaluation of ADPKD progression: a progress report. Clin J Am Soc Nephrol 2006; 1: 754-760
- Spithoven EM, Meijer E, Boertien WE: Tubular secretion of creatinine in ADPKD: consequences for cross-sectional and longitudinal perfomance of kidney function estimating equations. Am J Kidney Dis 2013; 62: 531-540
- Jafar TH, Stark PC, Schnid CH: The effect of angiotensin-converting-enzyme inhibitors on pregression of advanced PKD. Kidney Int 2005; 67: 265-271
- Wang D, Iversen J, Wilcox CS: Endothelial dysfunction and reduced nitric oxide in resistance arteries in ADPKD.Kidney Int 2003; 64: 1381-1388
- Nutahara K, Higashihara E, Hories S: Calcium channel blocker versus angiotensin II receptor blocker in ADPKD. Nephron Clin Pract 2005; 99: c18-23
- Thierbach R, Schulz TJ, Isken A, Voigt B, Mietzner G et al: Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum Mol Genet 2005; 14(24): 3857-3864
- Motoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O et al: Regulates mitochondrial respiration. Science 2006; Jun 16; 312(5780): 1650-1653
- Ristow M: Oxidative metabolism in cancer growth. Current Opinion in Clinical Nutrition and Metabolic care. 2006, Vol 9, n4, July: 339-345
- Weinhouse S: The Warburg hypothesis fifty yars later. Journal of Cancer Research and Clinical Oncology. 1976, vol 87, n2: 115-126
- 4th Annual CRISPR, Boston Nov 2023
- Savier E: Editing genomes with the bacterial immune system. 2013; Nature Publishing Group
- Charpentier E, Kaldy P: L’enzima che rivoluziona la genetica. Le Scienze n 772 Apr 2016; 28-35
- Moijca Francisco JM, Rodriguez-Valera F: The discovery of CRISPR in archea and bacteria. The FEBS Journal vol 283, n17 Sept 2016; 3162-3169